Senolytic drugs are agents that selectively induce apoptosis of senescent cells. These cells accumulate in many tissues with aging and at sites of pathology in multiple chronic diseases. In studies in animals, targeting senescent cells using genetic or pharmacological approaches delays, prevents, or alleviates multiple age-related phenotypes, chronic diseases, geriatric syndromes, and loss of physiological resilience. Among the chronic conditions successfully treated by depleting senescent cells in preclinical studies are frailty, cardiac dysfunction, vascular hyporeactivity and calcification, diabetes mellitus, liver steatosis, osteoporosis, vertebral disk degeneration, pulmonary fibrosis, and radiation-induced damage. Senolytic agents are being tested in proof-of-concept clinical trials. To do so, new clinical trial paradigms for testing senolytics and other agents that target fundamental aging mechanisms are being developed, because use of long-term endpoints such as lifespan or healthspan is not feasible. These strategies include testing effects on multimorbidity, accelerated aging-like conditions, diseases with localized accumulation of senescent cells, potentially fatal diseases associated with senescent cell accumulation, age-related loss of physiological resilience, and frailty. If senolytics or other interventions that target fundamental aging processes prove to be effective and safe in clinical trials, they could transform geriatric medicine by enabling prevention or treatment of multiple diseases and functional deficits in parallel, instead of one at a time.
A senolytic (from the words senescence and -lytic, "destroying") is among a class of small molecules under basic research to determine if they can selectively induce death of senescent cells and improve health in humans.[1] A goal of this research is to discover or develop agents to delay, prevent, alleviate, or reverse age-related diseases.[2][3] A related concept is "senostatic", which means to suppress senescence.
USP7 inhibitors
Inhibitors of USP7 (ubiquitin-specific processing protease 7)[15]
Senescence-specific killing compound 1: A gemcitabine (a cytotoxic chemotherapeutic) prodrug that is activated by lysosomal β-galactosidase (a common senescence marker)[27]
Target the enzyme kidney-type glutaminase 1 (GLS1). Senescent cells have a low pH due to their high lysosomal content and leaking lysosomal membranes. This low pH forms the basis of senescence-associated beta-galactosidase (SA-β-gal) staining of senescent cells. To help neutralize their low pH, senescent cells produce high levels of GLS1; inhibiting the activity of this enzyme exposes senescent cells to unsurvivably severe internal acidity, leading to cell death.[29]
FOXO4 binding to p53 protein retains it in the nucleus, which prevents it from interacting with mitochondria in the cytosol where it would activate caspases, leading to apoptosis (programmed cell death).[16] Instead, retention of p53 in the nucleus by FOXO4 promotes cellular senescence.[16] A peptide that binds with FOXO4 disrupts the p53-FOXO4 interaction, releasing p53 into the cytosol and triggering cell death.[16]
Crispr/Cas9 BIRC5 Gene Knockout. Crispr/Cas9 is used to trigger apoptosis in relation to a specified gene sequence such as a cancer gene sequence or damage marker sequences.[28]
Glycoprotein nonmetastatic melanoma protein B (GPNMB). A protein that enrich senescent cells studied as molecular target for a senolytic vaccine in mice.[30]
I haven't heard anyone classify rapamycin as a senolytic before. It is often compared and contrasted with senolytics though, and some of the more adventurous are taking rapamycin together with senolytics. Rapamycin is thought to slow aging by inhibiting mTOR and has the effect of preventing cells from becoming senescent, versus the senolytics which kill off senescent cells.
Senolytics are basically anti-cancer drugs, repurposed to kill senescent cells selectively. It is even more difficult to selectively kill senescent cells than to kill cancer cells. Based on lessons of cancer therapy, here I suggest how to exploit ...
www.ncbi.nlm.nih.gov
I've tried fisetin as a senolytic before in a 2 gram dose. It caused my joints to flare up, specifically the ones that were bothering me already. They may have felt better than usual afterwards. Or it might all have been placebo. Some have seen more dramatic effects though.
I've also used rapamycin before at a dose of 5 mg weekly. That was well tolerated except that it seemed to reduce my already low libido further, which was a deal breaker for me. I'll revisit that in the future when I fix my libido to the point where I can afford to lose a bit.
I take 4mg every other week. Some people notice weight loss when they start and I noticed this as well. I assume it was not muscle as I did not lose any strength. Beyond that it is hard to say what it is accomplishing given everything else I take and do. There is a researcher (whose name I'm blanking on) who showed that it extended life/healthspan in dogs.
I take 4mg every other week. Some people notice weight loss when they start and I noticed this as well. I assume it was not muscle as I did not lose any strength. Beyond that it is hard to say what it is accomplishing given everything else I take and do. There is a researcher (whose name I'm blanking on) who showed that it extended life/healthspan in dogs.
Rapamycin is not a senolytic compound. It is believed to have senomorphic effects, but this is poorly understood at this point. Most of the benefits of rapamycin are believed to be due to increased autophagy as result of MTOR inihibition. A senolytic compound kills senescent cells. A senomorphic compound in some way alters senescent cells or their secrections.
I haven't heard anyone classify rapamycin as a senolytic before. It is often compared and contrasted with senolytics though, and some of the more adventurous are taking rapamycin together with senolytics. Rapamycin is thought to slow aging by inhibiting mTOR and has the effect of preventing cells from becoming senescent, versus the senolytics which kill off senescent cells.
Senolytics are basically anti-cancer drugs, repurposed to kill senescent cells selectively. It is even more difficult to selectively kill senescent cells than to kill cancer cells. Based on lessons of cancer therapy, here I suggest how to exploit ...
www.ncbi.nlm.nih.gov
I've tried fisetin as a senolytic before in a 2 gram dose. It caused my joints to flare up, specifically the ones that were bothering me already. They may have felt better than usual afterwards. Or it might all have been placebo. Some have seen more dramatic effects though.
I've also used rapamycin before at a dose of 5 mg weekly. That was well tolerated except that it seemed to reduce my already low libido further, which was a deal breaker for me. I'll revisit that in the future when I fix my libido to the point where I can afford to lose a bit.
I was attempting to add too many things to one thread, I see where it was a conflict, so I removed the references to Rapamycin. I think that subject should have its own dedicated thread. Thank you for pointing that out.
Rapamycin is not a senolytic compound. It is believed to have senomorphic effects, but this is poorly understood at this point. Most of the benefits of rapamycin are believed to be due to increased autophagy as result of MTOR inihibition. A senolytic compound kills senescent cells. A senomorphic compound in some way alters senescent cells or their secretions.
Cellular senescence is a hallmark of aging, it is a permanent state of cell cycle arrest induced by cellular stresses. During the aging process, senescent cells (SCs) increasingly accumulate in tissues, causing a loss of tissue-repair capacity because of cell cycle arrest in progenitor cells and...
pubmed.ncbi.nlm.nih.gov
Emerging senolytic agents derived from natural products
Cellular senescence is a hallmark of aging, it is a permanent state of cell cycle arrest induced by cellular stresses. During the aging process, senescent cells (SCs) increasingly accumulate in tissues, causing a loss of tissue-repair capacity because of cell cycle arrest in progenitor cells and produce proinflammatory and matrix-degrading molecules which are known as the senescence-associated secretory phenotype (SASP), and thereby contribute to the development of various age-related diseases. Genetic evidence has demonstrated that clearance of SCs can delay aging and extend healthspan. Senolytics, small molecules that can selectively kill SCs, have been developed to treat various age-related diseases. In recent years, emerging natural compounds have been discovered to be effective senolytic agents, such as quercetin, fisetin, piperlongumine and the curcumin analog. Some of the compounds have been validated in animal models and have great potential to be pushed to clinical applications. In this review, we will discuss cellular senescence and its potential as a target for treating age-related diseases, and summarize the known natural compounds as senolytic agents and their applications.
Not to be confused with Autophagia.
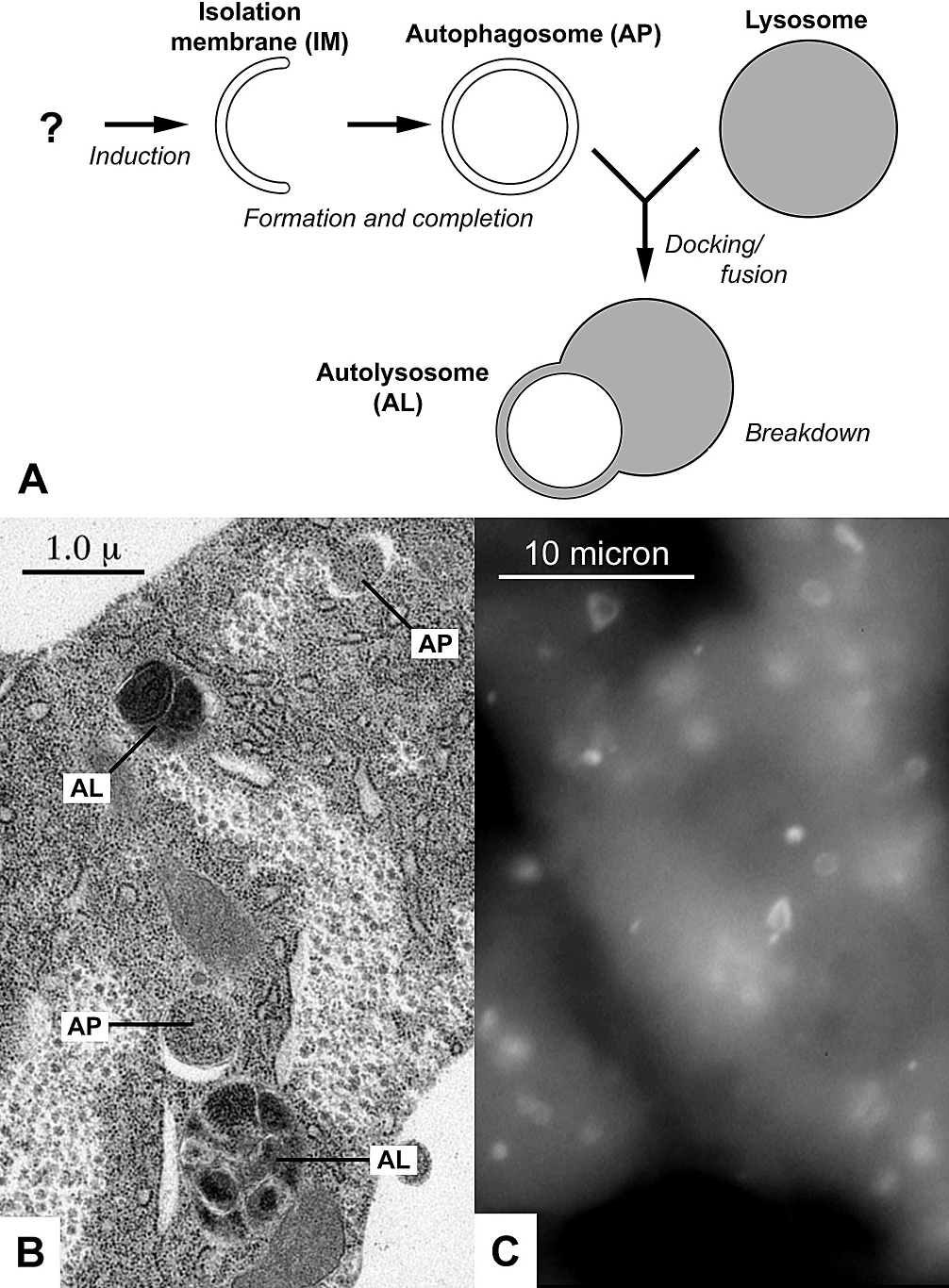
This article is about the cellular process. For other uses, see Autophagy (disambiguation). A Diagram of the process of autophagy, which produces the structures autophagosomes (AP), and autolysosomes (AL); B Electron micrograph of autophagic structures AP and AL in the fat body of a fruit fly larva; CFluorescently-labeled autophagosomes AP in liver cells of starved mice. Autophagy (or autophagocytosis; from the Ancient Greek αὐτόφαγος, autóphagos, meaning "self-devouring"[1] and κύτος, kýtos, meaning "hollow")[2] is the natural, conserved degradation of the cell that removes unnecessary or dysfunctional components through a lysosome-dependent regulated mechanism.[3] It allows the orderly degradation and recycling of cellular components.[4][5] Although initially characterized as a primordial degradation pathway induced to protect against starvation, it has become increasingly clear that autophagy also plays a major role in the homeostasis of non-starved cells.[6] Defects in autophagy have been linked to various human diseases, including neurodegeneration and cancer, and interest in modulating autophagy as a potential treatment for these diseases has grown rapidly.[6][7]
Four forms of autophagy have been identified: macroautophagy, microautophagy, chaperone-mediated autophagy (CMA), and crinophagy.[8] In macroautophagy (the most thoroughly researched form of autophagy), cytoplasmic components (like mitochondria) are targeted and isolated from the rest of the cell within a double-membrane vesicle known as an autophagosome,[9][10] which, in time, fuses with an available lysosome, bringing its specialty process of waste management and disposal; and eventually the contents of the vesicle (now called an autolysosome) are degraded and recycled. In crinophagy (the least well-known and researched form of autophagy), unnecessary secretory granules are degraded and recycled.[8]
In disease, autophagy has been seen as an adaptive response to stress, promoting survival of the cell; but in other cases, it appears to promote cell death and morbidity. In the extreme case of starvation, the breakdown of cellular components promotes cellular survival by maintaining cellular energy levels.
The word "autophagy" was in existence and frequently used from the middle of the 19th century.[11] In its present usage, the term autophagy was coined by Belgian biochemist Christian de Duve in 1963 based on his discovery of the functions of lysosome.[3] The identification of autophagy-related genes in yeast in the 1990s allowed researchers to deduce the mechanisms of autophagy,[12][13][14][15][16] which eventually led to the award of the 2016 Nobel Prize in Physiology or Medicine to Japanese researcher Yoshinori Ohsumi.[17]
Autophagy was first observed by Keith R. Porter and his student Thomas Ashford at the Rockefeller Institute. In January 1962 they reported an increased number of lysosomes in rat liver cells after the addition of glucagon, and that some displaced lysosomes towards the centre of the cell contained other cell organelles such as mitochondria. They called this autolysis after Christian de Duve and Alex B. Novikoff. However Porter and Ashford wrongly interpreted their data as lysosome formation (ignoring the pre-existing organelles). Lysosomes could not be cell organelles, but part of cytoplasm such as mitochondria, and that hydrolytic enzymes were produced by microbodies.[18] In 1963 Hruban, Spargo and colleagues published a detailed ultrastructural description of "focal cytoplasmic degradation", which referenced a 1955 German study of injury-induced sequestration. Hruban, Spargo and colleagues recognized three continuous stages of maturation of the sequestered cytoplasm to lysosomes, and that the process was not limited to injury states that functioned under physiological conditions for "reutilization of cellular materials", and the "disposal of organelles" during differentiation.[19] Inspired by this discovery, de Duve christened the phenomena "autophagy". Unlike Porter and Ashford, de Duve conceived the term as a part of lysosomal function while describing the role of glucagon as a major inducer of cell degradation in the liver. With his student Russell Deter, he established that lysosomes are responsible for glucagon-induced autophagy.[20][21] This was the first time the fact that lysosomes are the sites of intracellular autophagy was established.[3][22][23]
In the 1990s several groups of scientists independently discovered autophagy-related genes using the budding yeast. Notably, Yoshinori Ohsumi and Michael Thumm examined starvation-induced non-selective autophagy;[13][14][15] in the meantime, Daniel J. Klionsky discovered the cytoplasm-to-vacuole targeting (CVT) pathway, which is a form of selective autophagy.[12][16] They soon found that they were in fact looking at essentially the same pathway, just from different angles.[24][25] Initially, the genes discovered by these and other yeast groups were given different names (APG, AUT, CVT, GSA, PAG, PAZ, and PDD). A unified nomenclature was advocated in 2003 by the yeast researchers to use ATG to denote autophagy genes.[26] The 2016 Nobel Prize in Physiology or Medicine was awarded to Yoshinori Ohsumi,[17] although some have pointed out that the award could have been more inclusive.[27]
The field of autophagy research experienced accelerated growth at the turn of the 21st century. Knowledge of ATG genes provided scientists more convenient tools to dissect functions of autophagy in human health and disease. In 1999, a landmark discovery connecting autophagy with cancer was published by Beth Levine's group.[28] To this date, relationship between cancer and autophagy continues to be a main theme of autophagy research. The roles of autophagy in neurodegeneration and immune defense also received considerable attention. In 2003, the first Gordon Research Conference on autophagy was held at Waterville.[29] In 2005, Daniel J Klionsky launched Autophagy, a scientific journal dedicated to this field. The first Keystone Symposia Conference on autophagy was held in 2007 at Monterey.[30] In 2008, Carol A Mercer created a BHMT fusion protein (GST-BHMT), which showed starvation-induced site-specific fragmentation in cell lines. The degradation of betaine homocysteine methyltransferase (BHMT), a metabolic enzyme, could be used to assess autophagy flux in mammalian cells. Macro, micro, and Chaperone mediated autophagy are mediated by autophagy-related genes and their associated enzymes.[9][10][31][32][33] Macroautophagy is then divided into bulk and selective autophagy. In the selective autophagy is the autophagy of organelles; mitophagy,[34] lipophagy,[35] pexophagy,[36] chlorophagy,[37] ribophagy[38] and others. Macroautophagy is the main pathway, used primarily to eradicate damaged cell organelles or unused proteins.[39] First the phagophore engulfs the material that needs to be degraded, which forms a double membrane known as an autophagosome, around the organelle marked for destruction.[32][40] The autophagosome then travels through the cytoplasm of the cell to a lysosome in mammals, or vacuoles in yeast and plants,[41] and the two organelles fuse.[32] Within the lysosome/vacuole, the contents of the autophagosome are degraded via acidic lysosomal hydrolase.[42] Microautophagy, on the other hand, involves the direct engulfment of cytoplasmic material into the lysosome.[43] This occurs by invagination, meaning the inward folding of the lysosomal membrane, or cellular protrusion.[40] Chaperone-mediated autophagy, or CMA, is a very complex and specific pathway, which involves the recognition by the hsc70-containing complex.[40][44] This means that a protein must contain the recognition site for this hsc70 complex which will allow it to bind to this chaperone, forming the CMA- substrate/chaperone complex.[42] This complex then moves to the lysosomal membrane-bound protein that will recognise and bind with the CMA receptor. Upon recognition, the substrate protein gets unfolded and it is translocated across the lysosome membrane with the assistance of the lysosomal hsc70 chaperone.[31][32] CMA is significantly different from other types of autophagy because it translocates protein material in a one by one manner, and it is extremely selective about what material crosses the lysosomal barrier.[39] Mitophagy is the selective degradation of mitochondria by autophagy. It often occurs to defective mitochondria following damage or stress. Mitophagy promotes the turnover of mitochondria and prevents the accumulation of dysfunctional mitochondria which can lead to cellular degeneration. It is mediated by Atg32 (in yeast) and NIX and its regulator BNIP3 in mammals. Mitophagy is regulated by PINK1 and parkin proteins. The occurrence of mitophagy is not limited to the damaged mitochondria but also involves undamaged ones.[33] Lipophagy is the degradation of lipids by autophagy,[35] a function which has been shown to exist in both animal and fungal cells.[45] The role of lipophagy in plant cells, however, remains elusive.[46] In lipophagy the target are lipid structures called lipid droplets (LDs), spheric "organelles" with a core of mainly triacylglycerols (TAGs) and a unilayer of phospholipids and membrane proteins. In animal cells the main lipophagic pathway is via the engulfment of LDs by the phagophore, macroautophagy. In fungal cells on the other hand microplipophagy constitutes the main pathway and is especially well studied in the budding yeast Saccharomyces cerevisiae[47]. Lipophagy was first discovered in mice and published 2009.[48]
Targeted interplay between bacterial pathogens and host autophagy[edit]
Autophagy targets genus-specific proteins, so orthologous proteins which share sequence homology with each other are recognized as substrates by a particular autophagy targeting protein. There exists a complementarity of autophagy targeting proteins which potentially increase infection risk upon mutation. The lack of overlap among the targets of the 3 autophagy proteins and the large overlap in terms of the genera show that autophagy could target different sets of bacterial proteins from a same pathogen. On one hand, the redundancy in targeting a same genera is beneficial for robust pathogen recognition. But, on the other hand, the complementarity in the specific bacterial proteins could make the host more susceptible to chronic disorders and infections if the gene encoding one of the autophagy targeting proteins becomes mutated, and the autophagy system is overloaded or suffers other malfunctions. Moreover, autophagy targets virulence factors and virulence factors responsible for more general functions such as nutrient acquisition and motility are recognized by multiple autophagy targeting proteins. And the specialized virulence factors such as autolysins, and iron sequestering proteins are potentially recognized uniquely by a single autophagy targeting protein. The autophagy proteins CALCOCO2/NDP52 and MAP1LC3/LC3 may have evolved specifically to target pathogens or pathogenic proteins for autophagic degradation. While SQSTM1/p62 targets more generic bacterial proteins containing a target motif but not related to virulence.[49]
On the other hand, bacterial proteins from various pathogenic genera are also able to modulate autophagy. There are genus-specific patterns in the phases of autophagy that are potentially regulated by a given pathogen group. Some autophagy phases can only be modulated by particular pathogens, while some phases are modulated by multiple pathogen genera. Some of the interplay-related bacterial proteins have proteolytic and post-translational activity such as phosphorylation and ubiquitination and can interfere with the activity of autophagy proteins.[49]
Autophagy is executed by autophagy-related (Atg) genes. Prior to 2003, ten or more names were used, but after this point a unified nomenclature was devised by fungal autophagy researchers.[50] Atg or ATG stands for autophagy related. It does not specify gene or a protein.[50]
The first autophagy genes were identified by genetic screens conducted in Saccharomyces cerevisiae.[12][13][14][15][16] Following their identification those genes were functionally characterized and their orthologs in a variety of different organisms were identified and studied.[9][51] Today, thirty-six Atg proteins have been classified as especially important for autophagy, of which 18 belong to the core machinery[52]
In mammals, amino acid sensing and additional signals such as growth factors and reactive oxygen species regulate the activity of the protein kinasesmTOR and AMPK.[51][53] These two kinases regulate autophagy through inhibitory phosphorylation of the Unc-51-like kinases ULK1 and ULK2 (mammalian homologues of Atg1).[54] Induction of autophagy results in the dephosphorylation and activation of the ULK kinases. ULK is part of a protein complex containing Atg13, Atg101 and FIP200. ULK phosphorylates and activates Beclin-1 (mammalian homologue of Atg6),[55] which is also part of a protein complex. The autophagy-inducible Beclin-1 complex[56] contains the proteins PIK3R4(p150), Atg14L and the class III phosphatidylinositol 3-phosphate kinase (PI(3)K) Vps34.[57] The active ULK and Beclin-1 complexes re-localize to the site of autophagosome initiation, the phagophore, where they both contribute to the activation of downstream autophagy components.[58][59]
Once active, VPS34 phosphorylates the lipidphosphatidylinositol to generate phosphatidylinositol 3-phosphate (PtdIns(3)P) on the surface of the phagophore. The generated PtdIns(3)P is used as a docking point for proteins harboring a PtdIns(3)P binding motif. WIPI2, a PtdIns(3)P binding protein of the WIPI (WD-repeat protein interacting with phosphoinositides) protein family, was recently shown to physically bind ATG16L1.[60] Atg16L1 is a member of an E3-like protein complex involved in one of two ubiquitin-like conjugation systems essential for autophagosome formation. The FIP200 cis-Golgi-derived membranes fuse with ATG16L1-positive endosomal membranes to form the prophagophore termed HyPAS (hybrid pre-autophagosomal structure).[61] ATG16L1 binding to WIPI2[62] mediates ATG16L1's activity. This leads to downstream conversion of prophagophore into ATG8-positive phagophore[61] via a ubiquitin-like conjugation system.
The first of the two ubiquitin-like conjugation systems involved in autophagy covalently binds the ubiquitin-like protein Atg12 to Atg5. The resulting conjugate protein then binds ATG16L1 to form an E3-like complex which functions as part of the second ubiquitin-like conjugation system.[63] This complex binds and activates Atg3, which covalently attaches mammalian homologues of the ubiquitin-like yeast protein ATG8 (LC3A-C, GATE16, and GABARAPL1-3), the most studied being LC3 proteins, to the lipid phosphatidylethanolamine (PE) on the surface of autophagosomes.[64] Lipidated LC3 contributes to the closure of autophagosomes,[65] and enables the docking of specific cargos and adaptor proteins such as Sequestosome-1/p62.[66] The completed autophagosome then fuses with a lysosome through the actions of multiple proteins, including SNAREs[67][68] and UVRAG.[69] Following the fusion LC3 is retained on the vesicle's inner side and degraded along with the cargo, while the LC3 molecules attached to the outer side are cleaved off by Atg4 and recycled.[70] The contents of the autolysosome are subsequently degraded and their building blocks are released from the vesicle through the action of permeases.[71] Sirtuin 1 (SIRT1) stimulates autophagy by preventing acetylation of proteins (via deacetylation) required for autophagy as demonstrated in cultured cells and embryonic and neonatal tissues.[72] This function provides a link between sirtuin expression and the cellular response to limited nutrients due to caloric restriction.[73]
Autophagy has roles in various cellular functions. One particular example is in yeasts, where the nutrient starvation induces a high level of autophagy. This allows unneeded proteins to be degraded and the amino acids recycled for the synthesis of proteins that are essential for survival.[74][75][76] In higher eukaryotes, autophagy is induced in response to the nutrient depletion that occurs in animals at birth after severing off the trans-placental food supply, as well as that of nutrient starved cultured cells and tissues.[77][78] Mutant yeast cells that have a reduced autophagic capability rapidly perish in nutrition-deficient conditions.[79] Studies on the apg mutants suggest that autophagy via autophagic bodies is indispensable for protein degradation in the vacuoles under starvation conditions, and that at least 15 APG genes are involved in autophagy in yeast.[79] A gene known as ATG7 has been implicated in nutrient-mediated autophagy, as mice studies have shown that starvation-induced autophagy was impaired in atg7-deficient mice.[78]
Vesicular stomatitis virus is believed to be taken up by the autophagosome from the cytosol and translocated to the endosomes where detection takes place by a pattern recognition receptor called toll-like receptor 7, detecting single stranded RNA. Following activation of the toll-like receptor, intracellular signaling cascades are initiated, leading to induction of interferon and other antiviral cytokines. A subset of viruses and bacteria subvert the autophagic pathway to promote their own replication.[80]Galectin-8 has recently been identified as an intracellular "danger receptor", able to initiate autophagy against intracellular pathogens. When galectin-8 binds to a damaged vacuole, it recruits an autophagy adaptor such as NDP52 leading to the formation of an autophagosome and bacterial degradation.[81]
Autophagy degrades damaged organelles, cell membranes and proteins, and insufficient autophagy is thought to be one of the main reasons for the accumulation of damaged cells and aging.[82] Autophagy and autophagy regulators are involved in response to lysosomal damage, often directed by galectins such as galectin-3 and galectin-8.
One of the mechanisms of programmed cell death (PCD) is associated with the appearance of autophagosomes and depends on autophagy proteins. This form of cell death most likely corresponds to a process that has been morphologically defined as autophagic PCD. One question that constantly arises, however, is whether autophagic activity in dying cells is the cause of death or is actually an attempt to prevent it. Morphological and histochemical studies have not so far proved a causative relationship between the autophagic process and cell death. In fact, there have recently been strong arguments that autophagic activity in dying cells might actually be a survival mechanism.[83][84] Studies of the metamorphosis of insects have shown cells undergoing a form of PCD that appears distinct from other forms; these have been proposed as examples of autophagic cell death.[85] Recent pharmacological and biochemical studies have proposed that survival and lethal autophagy can be distinguished by the type and degree of regulatory signaling during stress particularly after viral infection.[86] Although promising, these findings have not been examined in non-viral systems.
Autophagy is essential for basal homeostasis; it is also extremely important in maintaining muscle homeostasis during physical exercise.[87][88] Autophagy at the molecular level is only partially understood. A study of mice shows that autophagy is important for the ever-changing demands of their nutritional and energy needs, particularly through the metabolic pathways of protein catabolism. In a 2012 study conducted by the University of Texas Southwestern Medical Center in Dallas, mutant mice (with a knock-in mutation of BCL2 phosphorylation sites to produce progeny that showed normal levels of basal autophagy yet were deficient in stress-induced autophagy) were tested to challenge this theory. Results showed that when compared to a control group, these mice illustrated a decrease in endurance and an altered glucose metabolism during acute exercise.[89]
Another study demonstrated that skeletal muscle fibers of collagen VI in knockout mice showed signs of degeneration due to an insufficiency of autophagy which led to an accumulation of damaged mitochondria and excessive cell death.[90] Exercise-induced autophagy was unsuccessful however; but when autophagy was induced artificially post-exercise, the accumulation of damaged organelles in collagen VI deficient muscle fibres was prevented and cellular homeostasis was maintained. Both studies demonstrate that autophagy induction may contribute to the beneficial metabolic effects of exercise and that it is essential in the maintaining of muscle homeostasis during exercise, particularly in collagen VI fibers.[89][88][90]
Work at the Institute for Cell Biology, University of Bonn, showed that a certain type of autophagy, i.e. chaperone-assisted selective autophagy (CASA), is induced in contracting muscles and is required for maintaining the muscle sarcomere under mechanical tension.[91] The CASA chaperone complex recognizes mechanically damaged cytoskeleton components and directs these components through a ubiquitin-dependent autophagic sorting pathway to lysosomes for disposal. This is necessary for maintaining muscle activity.[91][92]
Multiple guys on here are taking it. You can ask your doctor for a script, some will oblige. Empower Pharmacy sells it, you need to go through Defy. And the Anti Aging Store sells it with no prescription.
Multiple guys on here are taking it. You can ask your doctor for a script, some will oblige. Empower Pharmacy sells it, you need to go through Defy. And the Anti Aging Store sells it with no prescription.
There are a couple of good ******** groups where they discuss the questions you're asking. There is also lots of good info here: Rapamycin Longevity News
Fernando... If I ever give you a source, it's legit. I have used Anti Aging Systems for years.
There is no average age, people start when they learn about it and decide to try it. It is more effective the earlier you start however. There are multiple studies showing this.
Fernando... If I ever give you a source, it's legit. I have used Anti Aging Systems for years.
There is no average age, people start when they learn about it and decide to try it. It is more effective the earlier you start however. There are multiple studies showing this.
I have been on 2x1000mg a day but I lost my endurance(Kun Khmer/Muay Thai training) and workout capacity/volume. I had no common sides from it, no diarrea etc, but felt physically limited. A strange feeling of being limited by the amount of energy the body had stored or could generate on the...
I have been on 2x1000mg a day but I lost my endurance(Kun Khmer/Muay Thai training) and workout capacity/volume. I had no common sides from it, no diarrea etc, but felt physically limited. A strange feeling of being limited by the amount of energy the body had stored or could generate on the...
Check out Dr. Matt Kaberline on youtube he says he took it for frozen shoulder with amazing results. So I do look foward to more studies(human) in the future. It does give cold sores as a side effect.
Check out Dr. Matt Kaberline on youtube he says he took it for frozen shoulder with amazing results. So I do look foward to more studies(human) in the future. It does give cold sores as a side effect.